Reactivity in Chemistry
Coordination Chemistry
Part CC9. Ligand Field Stabilisation Energy
There is a variation on how to think about d orbital
splitting diagrams that can be useful in deciding how the d electrons are
configured in transition metal complexes. We can use the relative energy
levels of the d orbitals in a given complex to calculate whether the overall
energy would be higher or lower in a high-spin vs. a low-spin case, for example.
The calculation provides us with a value that is called the ligand field
stabilisation energy. Although we have been thinking of bonding in
transition metal complexes in terms of molecular orbital ideas, ligand field
stabilisation energy actually has its roots in a separate approach called crystal
field theory.
Origin Story
Crystal field theory was independently developed
around 1930 by German physicist
Hans Bethe and American
physicist John
Hasbrouck van Vleck; the two later became professors of physics at Cornell
and Harvard, respectively. It actually pre-dates the molecular orbital approach
that we have been using so far, but it reaches similar conclusions about
transition metal electron configurations.
Both scientists were interested in the magnetic
properties of metals and metal salts. They knew these properties were
related to unpaired electrons. Compounds with unpaired electrons are
attracted by magnetic fields, whereas compounds having only paired electrons are
not. They were interested in the factors that influenced the d electron
configuration of transition metal salts, because how the d electrons filled
could result in different numbers of unpaired electrons. Those differences
influenced how strongly the compounds interacted with magnetic fields.
Crystal field theory does not consider any bonding
interactions in transition metal compounds. It focusses only on the repulsion
between the electrons on an anion and the electrons on a metal cation. Of course,
all of these electrons have negative charges. As an anion approaches a
metal, the physicists reasoned, both sets of electrons would experience
repulsive forces that would increase their energy level.
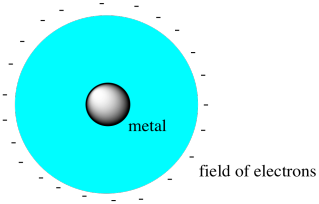
Figure CC9.1. A metal atom surrounded by a spherical electric field.
- Crystal field theory doesn't consider bonding interactions between ligands and metal atoms or ions.
- Crystal field theory imagines the repulsive interactions between metal electrons and the ligand electrons.
If the metal were surrounded by a spherical field of
electrons (physicists like to assume things are spherical because it makes the
math easier), then all of the electrons on the metal would be raised equally
high in energy by these repulsive forces.
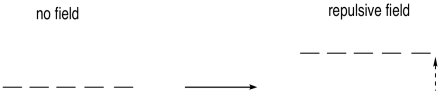
Figure CC9.2. The metal d electrons would be raised in energy by a spherical electric field.
But in a metal salt, electrons are not approaching
the metal from all directions. The geometry comes in
because of the fact that the cations and anions pack together in specific
arrays. For example, if a metal cation is sitting in an octahedral
hole, electrons would only approach from six
directions: at each end of the x, y, and z axes.
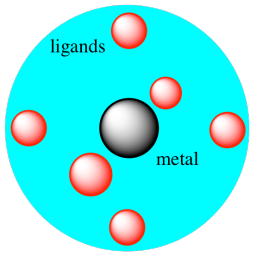
Figure CC9.3. A metal atom surrounded by an octahedral electric field.
In that case, only the electrons in d orbitals that
were aligned along the x, y and z axes would be raised significantly by
repulsion with the approaching electrons on the anions. The electrons in
the off-axis
orbitals would be lower in energy than they would have been in a perfectly spherical
field.
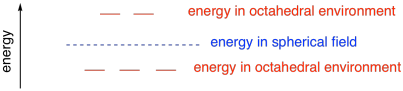
Figure CC9.4. The energy level of the d orbitals on an octahedral field
compared to a spherical electric field.
- Crystal field theory compares the energy of metal electrons in a specific coordination geometry
to what they would experience in a spherical electric field.
- Electrons in orbitals pointing directly at the ligands are raised significantly in energy
because of repulsion with the donor electrons.
- Electrons in orbitals between the ligands are lower in energy than they would be in a
spherical electric field.
Scientists later adapted this idea to octahedral
coordination complexes, in which the metal sits amidst six ligands rather than
getting packed among six anions.
That result leads to a picture that is pretty similar
to what we get from molecular orbital theory, at least as far as the d orbitals
are concerned. In fact, the true mathematical approach to molecular
orbital theory does take these electron-electron repulsions into account, but it
also factors in attractions between the electrons of the ligands and the nucleus
of the metal (and vice versa). The biggest difference is that molecular
orbital theory includes what happens to the kinetic energy of the ligand lone
pairs as they become shared with the metal (it goes down; that's a big part of
why the bonds form).
Application
You don't really have to appreciate where this
approach came from to be able to see how it is commonly used. If we take
the five d orbitals after they have been placed in an octahedral environment, we
see that they are at two different energy levels. The average energy level
is not, as it might first appear, halfway between these levels. That's
because there are three lower levels and two upper levels. The average is
two-fifths of the way up from the bottom three, or three-fifths of the way down
from the top two.
That average energy level of the five d orbitals is
called the "barycenter"; it is assigned a relative energy of zero. The
difference between the top level and the bottom level is the field splitting;
for an octahedral complex, this field splitting is given the symbol Δo; here,
Δ stands for the energy difference and o stands for octahedral.
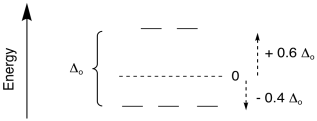
Figure CC9.5. The field splitting in an octahedral complex.
- The energy difference between the lower and higher energy d orbitals in an octahedral
complex is given the symbol Δo, "delta oh".
If the barycenter is at an energy level of zero, then the lower orbitals are
below zero. How far? They are two-fifths of the total Δo
below the barycenter. Any electrons in those orbitals are -0.4Δo below 0 in energy.
Electrons in the upper orbitals are +0.6Δo above zero in energy.
Suppose there are four electrons. The first three electrons sit in each of
the three lower orbitals, the ones we sometimes label the t2g
orbitals.. The fourth will either go into the upper level, which we
sometimes call the eg level, or else pair up with another electron at
the lower level.

Figure CC9.6. High spin and low spin cases in an octahedral electric field.
- Sometimes there are two different possible electron configurations.
- The complex will always adopt the configuration with lowest energy.
We can now calculate the energy difference between these two possible cases.
We can calculate what is called the ligand field stabilisation energy, LFSE
(sometimes called crystal field stabilisation energy, or CFSE).
It's just the sum of the energies of each of the electrons.
LFSE = [(0.6 x number of eg electrons) - (0.4 x number of t2g
electrons)]Δo
or if that's too much jargon, LFSE = [0.6 x # upper e-) - (0.4 x # lower e-)]Δo
On the left hand side, the high-spin case, that's:
LFSE = [(0.6 x 1) - (0.4 x 3)]Δo = [0.6 - 1.2]Δo = -0.6Δo
On the right hand side, that's
LFSE = [(0.6 x 0) - (0.4 x 4)]Δo = -1.6Δo
So far, it certainly seems like the low-spin case is at lower energy.
Remember, though, that it requires that we place two
electrons together into the same orbital.

Figure CC9.7. The energy trade-offs between high spin and low spin.
- Putting electrons in a higher-energy orbital costs energy.
- Putting two electrons in the same orbital also costs energy.
Like charges repel. Putting these
two electrons so close together is going to cost some energy. But how
much?

Figure CC9.8. Pairing energy occurs between two electrons in the same orbital.
This repulsion between a pair of electrons in one orbital is called the pairing
energy (PE). For each pair of electrons that occupy the same orbital, that
energy must be added to take that repulsion into account. As a result, a
calculation of the overall stabilisation energy includes both the ligand field
stabilisation energy and the pairing energy.
SE = LFSE + PE
If there were two sets of paired electrons, we would add 2PE; if there were
three sets of paired electrons, we would add 3PE, and so on.
Overall, deciding quantitatively whether a complex will be high spin or low spin
is the most useful application of an LFSE calculation. However, you do
have to know the values of the parameters (the field splitting and the pairing
energy) for a definitive decision.
Problem CC9.1.
Use SE calculations to
determine stabilisation in both high spin and low spin cases. Just leave your
answer expressed in terms of Δo and PE.
a. Fe+2
b. Co+2
c. Co+3
d. Mn+2
e. Ti+3
The Magnitude of Δo and PE
Taking the pairing energy into account in this case suggests that the high-spin
case is favoured. It cost less energy to jump the gap and put an electron
in a high-lying eg orbital than it did to pair electrons in a
low-lying t2g
orbital.
Right now, let's think about Δo. How much energy is it? How big is the gap between the d orbitals?
And how do we know? Well, we can easily measure this gap using a simple
spectrophotometer. A spectrophotometer measures how much light is absorbed
by a sample. Furthermore, it measures what specific colours of light are
absorbed by the sample. As it happens, the absorption of ultraviolet and
visible light by a material is associated with electrons in that material
becoming promoted to a higher energy level. Shine a light on a transition
metal complex, and an electron may jump the gap.
So all we have to do is shine different colours of light on the complex and
see whether it absorbs one of the colours.
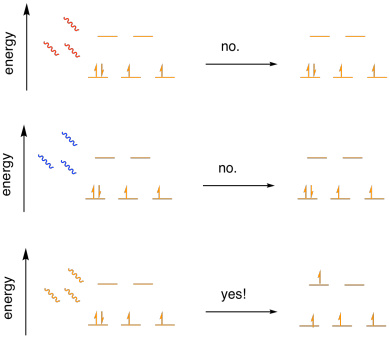
Figure CC9.9. The Goldilocks principal in quantum mechanics: the energy of a photon has to be just right
to excite an electron to the next level (not too much, not too little).
This experiment is quite reproducible. A specific compound will always
absorb the same colours of light. That's why specific materials have specific
colours; the colour we see represents the light that is not absorbed by
the material.
Different colours of light have different amounts of energy. Blue light
has higher energy than orange light, which has higher energy than red light.
Max Planck and Albert Einstein worked out that the energy of a photon (a
"particle" or "packet" of light) is directly proportional to the frequency at
which the photon oscillates or vibrates. Bue light has a higher frequency
than red light, so it has higher energy.
Planck and Einsten expressed this idea with an equation,
E = hν
in which the Greek letter ν, pronounced "noo", stands for the frequency (some people just use f for frequency) and h stands
for a sort of conversion factor called Planck's constant.
Blue light has too much energy to promote an electron to the next energy level
in this case. It would send the electron way past the next level, and
quantum mechanics doesn't allow that sort of thing. Red light doesn't have
enough energy for the electron to get there. Orange light, in this case,
is just right. It has exactly the right amount of energy to jump the gap.
Figure CC9.10. The energy of a photon is related to its wavelength and colour.
- We can tell the energy difference between orbitals if a photon promotes an electron from one orbital to another.
- The energy difference between the orbitals is equal to the energy of the photon
So, if we know the frequency of orange light, we know how much energy there is
in an orange photon, and we know how big the field splitting is between the d
orbitals.
Now, it turns out that historically people have most often described visible
light in terms of wavelength rather than frequencies. The wavelength is just the
distance from one "peak" to the next as the wave of the photon rolls along. The
higher the frequency, the closer these peaks are together, and the shorter the
wavelength. Blue light has a shorter wavelength, around 400 nm, than red
light, around 700 nm. A nanometer (nm) is 10-9 metres (m); a
metre is around a yard.
Orange light has a wavelength of around 600 nm, or 600 x 10-9 m, or 6
x 10-7 m. For reasons we won't get into, spectroscopists in the
past (people who measure the interaction of light and matter) sometimes
preferred to work in centimetres, cm; there are 100 cm in 1 m, so orange light
has a wavelength of 6 x 10-5 cm. Now, because they knew there
was an inverse relationship between wavelength and energy (the longer the
wavelength, the lower the energy; the shorter the wavelength, the higher the
energy), they simply took the reciprocal of the wavelength in centimeters to get
a number in cm-1, which they called wavenumbers. They used this
as a unit of energy. An orange photon has an energy of 1/0.00006 cm =
16,000 cm-1.
So the gap, the field splitting, Δo in this complex is 16,000 cm-1.
Let's go back to our comparison between the high-spin and low-spin case. For
high spin, that's:
LFSE = [(0.6 x 1) - (0.4 x 3)]Δo = [0.6 - 1.2]Δo = -0.6Δo =
-0.6 x 16,000 cm-1 = -9,600 cm-1
For the low-spin case, that's
LFSE = [(0.6 x 0) - (0.4 x 4)]Δo = -1.6Δo =
-1.6 x 16,000 cm-1 = -25,600 cm-1
That means that the low-spin case is lower in energy, by 14,000
cm-1.
However, we still need to include the pairing energy. Like the field
splitting, the pairing energy varies from one complex to another. 20,000
cm-1 is
a ballpark estimate of a typical pairing energy.
If we use this average value for PE in the example we were discussing above, for
the high-spin case:
SE = LFSE + PE = -9,600 + 0 cm-1 = -9,600 cm-1
For the low-spin case,
SE =
LFSE + PE = -25,600 + 20,000 cm-1 = -5,600 cm-1
Taking the pairing energy into account in this case suggests that the high-spin
case is favoured. It cost less energy to jump the gap and put an electron
in a high-lying eg orbital than it did to pair electrons in a
low-lying t2g
orbital.
Let's take a look at some real examples of field splitting values to get an idea
of how large they are, and what factors they depend on. For example, we can look
at the charge on the metal ion.
|
Table C9.1. Field Splitting Values, Δo, in Hexaaquo
Complexes of Differing Chargesa |
| Metal Complex |
[Mn(OH2)6]2+ |
[Mn(OH2)6]3+ |
[Fe(OH2)6]2+ |
[Fe(OH2)6]3+ |
[Co(OH2)6]2+ |
[Co(OH2)6]3+ |
| Δo (cm-1) |
7,800 |
21,100 |
10,400 |
13,800 |
9.300 |
18,300 |
| a) Holleman, A. F.; Wiberg, E.;
Wiberg, N. Inorganic Chemistry, 34th Ed. Academic Press: Berlin, 2001. |
|
|
In this case, it looks like increased charge on the metal ion results in an
increased field splitting. The same thing happens in all three examples:
manganese, iron, and cobalt. As the metal ion becomes more charged,
attraction of the d electrons toward the nucleus increases. The
lower-lying t2g level is more strongly attracted to the nucleus
because it is a little closer; consequently, it drops a little further than the
eg level, and the gap gets bigger.
Charge on the metal ion is one of two key factors that influence the size of
the field splitting. The other factor is the period or row in the periodic
table.
|
Table C9.2. Table of Field Splitting Values, Δo, in Hexaammine Complexes of Group 9a,b |
| Metal Complex |
[Co(NH3)6]3+ |
[Rh(NH3)6]3+ |
[Ir(NH3)6]3+ |
| Δo (cm-1) |
21,500 |
33,100 |
41,100 |
| a) Holleman, A. F.; Wiberg, E.;
Wiberg, N. Inorganic Chemistry, 34th Ed. Academic Press: Berlin, 2001. b) Miessler, G. L.; Tarr, D. A.
Inorganic Chemistry, 4th Ed. Pearson, 2010. |
Moving from one row to the next in group 9 of the periodic table, we see that
the field splitting increses by about 10,000 cm-1 each row.
In general:
- Δo increases with charge on the metal
- Δo increases with the period in the periodic table
The metal ion is not the only factor that affects the field splitting.
The ligands also play an important role, as seen in the table below.
|
Table C9.3. Field Splitting Values, Δo, in Assorted Complexesa |
| Metal Complex |
[CrCl6]3- |
[CrF6]3- |
[Fe(OH2)6]2+ |
[Fe(CN)6]4- |
[Co(OH2)6]3+ |
[Co(NH3)6]3+ |
[Co(CN)6]3- |
| Δo (cm-1) |
13,300 |
15,300 |
10,400 |
33,200 |
18,300 |
23,000 |
33,700 |
| a) Holleman, A. F.; Wiberg, E.;
Wiberg, N. Inorganic Chemistry, 34th Ed. Academic Press: Berlin, 2001. |
|
If we sort these ligands into three broad categories, we can form a trend for
these effects. It may be easiest to start with the three entries on the
right, involving cobalt. The oxygen donor in water has two lone pairs,
whereas the nitrogen donor in ammonia has only one. The extra lone pair
allows water to act as a π donor; the lone pair can donate to an orbital on
the metal to form a pi bond. In this case, it looks like the π donor (water)
results in a smaller Δo than the normal σ donor (ammonia).
The cyanide ligand has a donor atom that is also participating in a π bond within the ligand;
there is a triple bond between the C and N of the cyanide. That means
there is also an associated antibonding orbital, π*. That antibinding
orbital raises the possibility of back-bonding from the metal. The metal
can donate into the π* orbital to make a pi bond. Cyanide is a π-acceptor. Its
field splitting is much larger than the sigma donor.
Those conclusions can be confirmed by looking at the entries for iron in the
middle of the table. Cyanide is a π-acceptor whereas water is a π-donor.
The field splitting in the aquo complex should be much smaller than the field
splitting in the cyano complex, and it is.
The two entries for chromium on the left both show halides, which are π-donors.
They illustrate a factor that can be used to predict field strength between two
ligands from the same group, such as two π-donors or two π-acceptors.
In general, the more basic the ligand, the greater the field splitting.
Fluoride ion is more basic than chloride ion (because chloride is a more stable
anion than fluoride) so it results in a slightly greater value of Δo.
In order to thoroughly estimate the stabilisation energy, we also need
reliable values for the pairing energy. Pairing energies can be calculated
for free metal ions; correction factors can be applied to arrive at a
corresponding value in a complex. In general, the values in coordination
complexes are somewhat lower than the values in free metal ions. In the
table below, we have made a general estimate that the value in the complex is
about 20% lower than in the free metal ion.
|
Table C9.4. Pairing Energies, PE, in Free Ions, with Estimated PE in Complexes |
| Metal Ion |
Mn2+ |
Mn3+ |
Fe2+ |
Fe3+ |
Co3+ |
Ru3+ |
| PE, free ion (cm-1) |
25,500 |
28,000 |
17,700 |
30,100 |
21,100 |
~15,000b |
| PE, complex (est., cm-1) |
20,400 |
22,900 |
14,200 |
24,100 |
16,900 |
~12,000b |
| a) Holleman, A. F.; Wiberg, E.;
Wiberg, N. Inorganic Chemistry, 34th Ed. Academic Press: Berlin, 2001. b) Estimate. |
|
|
|
If we compare the pairing energies of the 2+ ions to those of the 3+ ions, we
see that it is always lower in the more highly charged ion. That's the
opposite of the trend in Δo. Pairing energy is largely dependent on the size of the ion.
The smaller the ion, the smaller the orbital, and the more repulsion between two
electrons in the same orbital. Because a Mn3+ ion is smaller
than a Mn2+ ion (Coulomb's Law says there is greater attraction for
the electrons in the former case, so the ion shrinks), a pair of electrons on Mn3+
is closer together than a pair of electrons on Mn2+.
In general, pairing energies also get smaller upon moving down a column in
the periodic table. Second row transition metals are larger than first-row
transition metals, so the pairing energy is smaller in the second row than in
the first. Third row transition metals are about the same size as first
row metals, so ther pairing energies are similar to those of the second row.
That's because of a phenomenon called "the lanthanide contraction": third row
transition metals contain an extra set of protons in their nuclei because of the
f block elements before them; consequently they are a little smaller than might
be expected.
- Pairing energies generally increase if charge increases on an ion.
- An ion contracts if it becomes more positive, so the repulsion between electrons increases.
- Generally, pairing energy is lower for second and third row transition metals than for first row transition metals.
- Second and third row transition metals are larger than in the first row so there is more room for the electrons and therefore less repulsion.
Problem CC9.2.
Compare pairing energies to field splitting values to
determine whether the following complexes would be high-spin or low spin.
a) Mn(OH2)62+
b) Fe(CN)64- c)
Co(NH3)63+
Other Geometries
Crystal field theory has also been applied to other geometries of
coordination compounds. Once
again, in terms of the d orbital splitting diagram, the results are similar to
what we see from molecular orbital theory. For tetrahedral geometry, which
is the most common geometry when the coordination number is four, we again get a
set of two high-lying orbitals and three lower ones.
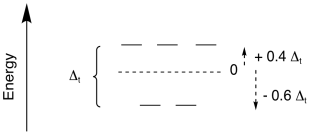
Figure CC9.11. The d orbital splitting in a tetrahedral field.
The overall splitting is expressed as Δt; the t stands for tetrahedral.
However, sometimes it is useful to compare different geometries. In
crystal field theory, it can be shown that the amount of splitting in a
tetrahedral field is much smaller than in an octahedral field. In general, Δt = 4/9 Δo.
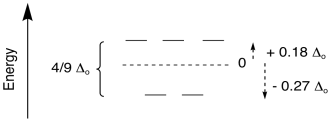
Figure CC9.12. The comparison of the magnitude of splitting in a tetrahedral field
to an octahedral field.
We wouldn't usually use crystal field theory to decide whether a metal is more likely to
adopt a tetrahedral or an octahedral geometry. In most cases, the outcome is more
strongly dependent on factors other than the d orbital energies. For
example, maybe a complex would be too crowded with six ligands, so it only binds
four; it becomes tetrahedral rather than octahedral. Or maybe a metal does
not have enough electrons in its valence shell, so it binds a couple more
ligands; it becomes octahedral rather than tetrahedral.
However, this comparison between Δo and
Δt does help to explain why tetrahedral complexes are much more likely
to adopt high-spin configurations than are octahedral complexes. The
splitting is smaller in tetrahedral geometry, so pairing energy is more likely
to become the deciding factor there than it is in octahedral cases.
Problem CC9.3.
Tetrahedral complexes are pretty common for high-spin d6 metals, even though the
18-electron rule suggests octahedral complexes should form. In contrast,
low-spin d6 complexes do not usually form tetrahedral complexes. Use
calculations of stabilisation energies to explain why.
Crystal field theory has also been used to determine the splittings between the
orbitals in a square planar geometry. That is a much more complicated
case, because there are four different levels. Once again, the energy
levels are often expressed in terms of Δo.
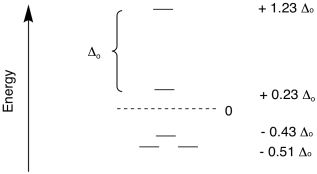
Figure CC9.13. The d orbital splitting in a square planar field.
We could use a calculation of stabilisation energy to predict whether a
particular complex is likely to adopt a tetrahedral or a square planar geometry.
Both geometries are possible, so it would be useful to be able to predict which
geometry occurs in which case. However, just as in the case of comparing
octahedral and tetrahedral gemoetries, there is another factor that is often
more important.
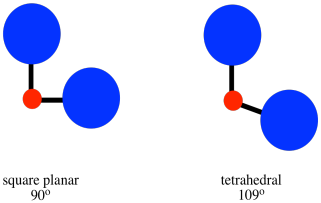
Figure CC9.14. Steric crowding can influence whether a complex is square planar or tetrahedral.
That factor is steric crowding. In a tetrahedron, all the ligands are 109o
from each other. In a square planar geometry, the ligands are only 90o
away from each other. Tetrahedral geomety is always less crowded than
square planar, so that factor always provides a bias toward tetrahedral
geometry. As a result, we might expect square planar geometry to occur
only when sterics is heavily outweighed by ligand field stabilisation energy.
Problem CC9.4.
The most common examples of square planar complexes have metals that are d8.
Use calculations of the stabilisation energy to explain why if the complex is:
(a) low-spin
(b) high-spin
This site was written by Chris P. Schaller, Ph.D., College of Saint Benedict / Saint John's
University (retired) with other authors as noted on individual pages. It is freely
available for educational use.

Structure & Reactivity in Organic, Biological and Inorganic Chemistry by
Chris Schaller is licensed under a Creative
Commons Attribution-NonCommercial 3.0 Unported License.
Send corrections to cschaller@csbsju.edu
This material is based upon work supported by the National Science Foundation
under Grant No. 1043566.
Any opinions, findings, and conclusions or recommendations expressed in this
material are those of the author(s) and do not necessarily reflect the views of
the National Science Foundation.
Navigation:
Back to Coordination Chemistry Index
Back to Reactivity
Back to Web Materials on Structure & Reactivity in Chemistry